Disease ー Doyne Honeycomb Retinal Dystrophy
Doyne Honeycomb Retinal Dystrophy, also known as Malattia Leventinese, is a genetic disorder characterized by early-onset macular degeneration leading to vision loss․ The condition primarily affects the central vision due to damage to the Retinal Pigment Epithelium (RPE) and photoreceptors․
Overview of Doyne Honeycomb Retinal Dystrophy
Doyne Honeycomb Retinal Dystrophy, or Malattia Leventinese, is a rare hereditary condition that falls under the category of macular dystrophies․ It is characterized by the formation of yellowish deposits under the retina which resemble a honeycomb pattern, hence its name․ The condition usually manifests in early adulthood and progresses over time, leading to significant visual impairment․
Individuals affected by Doyne Honeycomb Retinal Dystrophy often experience central vision problems, making tasks like reading, driving, and recognizing faces challenging․ The progressive deterioration of the macula, the central part of the retina responsible for detailed vision, results in a gradual decline in visual acuity․
While Doyne Honeycomb Retinal Dystrophy primarily affects the macula, it can also lead to complications such as choroidal neovascularization, where abnormal blood vessels grow under the retina․ This further exacerbates vision loss and can lead to more severe visual impairment if left untreated․
Understanding the early signs and symptoms of Doyne Honeycomb Retinal Dystrophy is crucial for prompt diagnosis and management․ Genetic testing plays a significant role in confirming the presence of mutations associated with the condition, allowing for targeted treatment approaches․ As research continues to advance, new therapeutic strategies are being developed to address the underlying mechanisms of Doyne Honeycomb Retinal Dystrophy and improve outcomes for affected individuals․
Understanding the Retina and Its Role in Vision
The retina is a complex layer of neural tissue located at the back of the eye that plays a vital role in the visual process․ Composed of several cell types, the retina is responsible for converting light signals into electrical impulses that can be interpreted by the brain․ Photoreceptor cells, namely rods and cones, capture light and transmit signals to neighboring retinal cells for processing and eventual transmission to the brain via the optic nerve․
The macula, a specialized area at the center of the retina, is crucial for detailed and central vision․ It contains a high concentration of cone photoreceptors, which are essential for tasks requiring visual acuity, such as reading and recognizing faces․ The integrity of the macula is essential for clear and sharp vision, especially in well-lit conditions․
In conditions like Doyne Honeycomb Retinal Dystrophy, damage to the macula can result in significant visual impairment․ The formation of cholesterol deposits known as drusen under the retina interferes with normal retinal function, leading to the progressive loss of central vision․ As the disease advances, the ability to perceive fine details and distinguish colors diminishes, impacting daily activities that rely on precise vision․
Understanding the intricate structure and function of the retina is essential in comprehending how diseases like Doyne Honeycomb Retinal Dystrophy disrupt vision․ Research into retinal physiology and pathology continues to provide insights into potential treatment strategies aimed at preserving retinal health and improving visual outcomes for individuals affected by retinal conditions․
Genetic Basis of Doyne Honeycomb Retinal Dystrophy
Doyne Honeycomb Retinal Dystrophy is primarily caused by mutations in the gene encoding the protein fibulin-3٫ also known as EFEMP1․ Fibulin-3 is essential for the maintenance of the extracellular matrix within the retina٫ contributing to its structural integrity and normal function․ Mutations in the EFEMP1 gene disrupt the production or function of fibulin-3٫ leading to the formation of characteristic drusen deposits under the macula․
The inheritance pattern of Doyne Honeycomb Retinal Dystrophy is typically autosomal dominant, meaning that a single altered copy of the EFEMP1 gene is sufficient to cause the condition․ In some cases, the disease may also be inherited in an autosomal recessive manner, requiring mutations in both copies of the EFEMP1 gene to manifest symptoms․
Genetic testing plays a crucial role in diagnosing Doyne Honeycomb Retinal Dystrophy by identifying specific mutations in the EFEMP1 gene․ Understanding the genetic basis of the disease not only aids in confirming diagnosis but also provides valuable information for genetic counseling and family planning․
Ongoing research aims to elucidate the intricate mechanisms through which EFEMP1 gene mutations contribute to the development and progression of Doyne Honeycomb Retinal Dystrophy․ By unraveling the genetic underpinnings of the disease٫ researchers strive to discover novel therapeutic targets that may slow or halt the retinal degeneration associated with this hereditary condition․
Early Onset and Progression of the Disease
Doyne Honeycomb Retinal Dystrophy typically presents in early adulthood, although the age of onset can vary among affected individuals․ Early symptoms may include blurred or distorted central vision, difficulty reading or recognizing faces, and sensitivity to bright light․ As the disease progresses, visual impairment worsens, leading to significant challenges in daily activities that rely on clear central vision․
The progression of Doyne Honeycomb Retinal Dystrophy is characterized by the gradual accumulation of drusen deposits beneath the macula․ These yellowish lesions disrupt the normal architecture of the retina, affecting the function of the Retinal Pigment Epithelium (RPE) and photoreceptor cells․ As a result, the macula deteriorates, leading to a decline in visual acuity and color vision․
Over time, individuals with Doyne Honeycomb Retinal Dystrophy may experience central scotomas, or blind spots in the central visual field, which further impede their ability to discern fine details․ The progression of the disease may vary in speed and severity among patients, with some experiencing more rapid deterioration of central vision than others․
Early detection and regular monitoring of the disease are crucial in managing Doyne Honeycomb Retinal Dystrophy․ By tracking changes in retinal structure and function over time, healthcare providers can implement interventions aimed at preserving remaining vision and improving quality of life for individuals affected by this genetic disorder․
Impact on Vision⁚ Vision Loss and Visual Impairment
Doyne Honeycomb Retinal Dystrophy exerts a profound impact on vision, leading to significant vision loss and visual impairment in affected individuals․ As the disease progresses, the accumulation of drusen deposits under the macula disrupts the normal function of the retina, particularly the central vision necessary for tasks like reading, driving, and recognizing faces․
Patients with Doyne Honeycomb Retinal Dystrophy often experience a gradual decline in visual acuity, with central vision being particularly affected․ The loss of central vision can manifest as blurriness, distortion, or blind spots in the central visual field, making it challenging to perform activities that require detailed vision or color discrimination․
Color vision may also be impaired in individuals with Doyne Honeycomb Retinal Dystrophy, further impacting the ability to perceive and differentiate colors accurately․ This can affect day-to-day activities such as selecting clothing, identifying traffic signals, or enjoying the visual arts․
The visual impairment caused by Doyne Honeycomb Retinal Dystrophy can have a profound impact on the quality of life of affected individuals․ Managing the challenges associated with vision loss requires a multidisciplinary approach involving ophthalmologists, low vision specialists, occupational therapists, and other healthcare professionals to optimize visual function and enhance independence and participation in daily activities․
Mechanism of Retinal Damage in Doyne Honeycomb Retinal Dystrophy
The mechanism of retinal damage in Doyne Honeycomb Retinal Dystrophy is primarily attributed to the accumulation of drusen deposits under the macula․ These yellowish deposits, composed of lipids and proteins, accumulate between the Retinal Pigment Epithelium (RPE) and Bruch’s membrane, disrupting the normal flow of nutrients and waste products within the retina․
The presence of drusen interferes with the exchange of oxygen, nutrients, and metabolic waste between the RPE and the underlying blood vessels, leading to oxidative stress and inflammation in the retinal layers․ The compromised function of the RPE, which is essential for supporting the photoreceptors and maintaining retinal health, contributes to the progressive degeneration of the macula․
In addition to the mechanical disruption caused by drusen accumulation, the presence of cholesterol deposits and lipofuscin within the retina further exacerbates retinal damage in Doyne Honeycomb Retinal Dystrophy․ These lipids and pigmented compounds can induce cellular toxicity, trigger inflammatory responses, and impair the function of retinal cells involved in the visual pathway․
Over time, the structural alterations and dysfunction of the macula due to drusen and lipid deposition result in the loss of photoreceptor cells and the deterioration of central vision․ The progressive nature of retinal damage in Doyne Honeycomb Retinal Dystrophy necessitates early detection, close monitoring, and targeted interventions to mitigate vision loss and preserve visual function for as long as possible․
Hereditary Nature of Doyne Honeycomb Retinal Dystrophy
Doyne Honeycomb Retinal Dystrophy is recognized for its inheritance pattern, which points to its hereditary nature․ The condition is predominantly passed down through generations in an autosomal dominant manner, meaning that a single copy of the mutated gene is adequate to cause the disorder․ In some cases, Doyne Honeycomb Retinal Dystrophy may exhibit autosomal recessive inheritance, requiring mutations in both copies of the gene for symptoms to manifest․
The genetic predisposition to Doyne Honeycomb Retinal Dystrophy is linked to mutations in the EFEMP1 gene, encoding the fibulin-3 protein crucial for maintaining retinal health․ Variations in the EFEMP1 gene can disrupt the structural integrity of the retina, leading to the formation of characteristic drusen deposits under the macula and subsequent vision loss․
Family history plays a significant role in understanding the hereditary nature of Doyne Honeycomb Retinal Dystrophy․ Individuals with affected family members have an increased risk of inheriting the genetic mutation associated with the condition․ Genetic counseling and testing are essential for assessing the likelihood of passing on the mutation to future generations and for understanding the implications of the disease within families․
By unraveling the hereditary basis of Doyne Honeycomb Retinal Dystrophy, researchers and healthcare professionals can provide tailored interventions and support for affected individuals and their families․ Early identification of at-risk individuals and proactive management strategies are crucial in addressing the genetic complexities of this inherited retinal disorder․
Mutations and Cholesterol Deposits in the Retina
Mutations in the EFEMP1 gene play a pivotal role in the pathogenesis of Doyne Honeycomb Retinal Dystrophy․ These genetic alterations disrupt the production or function of fibulin-3, a protein crucial for maintaining the extracellular matrix of the retina․ The presence of mutated fibulin-3 leads to the accumulation of drusen deposits under the macula—a hallmark feature of the disease․
Cholesterol deposits also contribute to the retinal pathology seen in Doyne Honeycomb Retinal Dystrophy․ These lipid-rich deposits accumulate between the Retinal Pigment Epithelium (RPE) and Bruch’s membrane, affecting the normal exchange of nutrients and waste products in the retina․ The combination of drusen and cholesterol deposits disrupts retinal function and leads to progressive vision loss․
The formation of drusen and cholesterol deposits not only interferes with nutrient transport within the retina but also induces inflammatory responses and oxidative stress in the retinal layers․ The presence of these deposits can trigger cellular damage, exacerbating the degenerative process and contributing to the impairment of retinal cells critical for vision․
Understanding the interplay between mutations in the EFEMP1 gene, fibulin-3 dysfunction, and the accumulation of cholesterol deposits is crucial in elucidating the mechanisms underlying retinal damage in Doyne Honeycomb Retinal Dystrophy․ Targeted research efforts aimed at addressing these molecular pathways hold promise for developing novel therapeutic strategies to counteract the degenerative processes and preserve visual function in individuals affected by this hereditary retinal disorder․
Involvement of Blood Vessels and Central Vision
In Doyne Honeycomb Retinal Dystrophy, the pathology extends beyond the retinal layers to involve the blood vessels supplying the retina․ The accumulation of drusen and cholesterol deposits under the macula can lead to changes in the underlying blood vessels, disrupting the normal flow of oxygen and nutrients to the retinal tissues․
The involvement of blood vessels in Doyne Honeycomb Retinal Dystrophy contributes to the development of complications such as choroidal neovascularization, where abnormal blood vessels grow beneath the retina․ These new vessels are fragile and prone to leaking, leading to further damage to the macula and exacerbating vision loss․
The impact on blood vessels in the macular region directly affects central vision, which relies heavily on the healthy functioning of the macula․ Changes in blood flow, oxygen delivery, and nutrient exchange due to vascular alterations can impair the metabolic support required by the photoreceptor cells in the macula, leading to their dysfunction and eventual degeneration․
The intricate relationship between blood vessels, metabolic support, and central vision underscores the complex etiology of vision loss in Doyne Honeycomb Retinal Dystrophy․ Understanding the interplay between vascular changes and retinal damage is crucial for developing targeted interventions that address not only the structural changes in the retina but also the vascular abnormalities that contribute to disease progression․
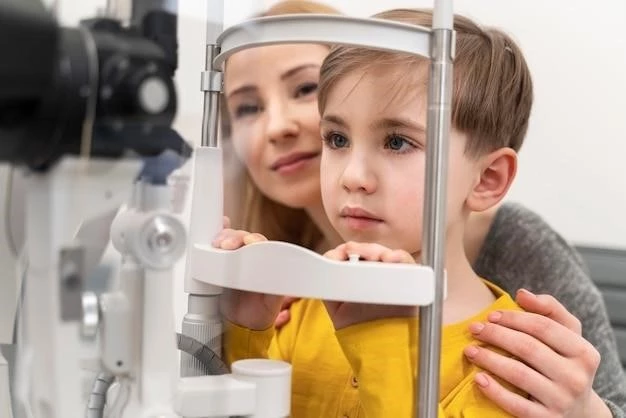
Diagnosis and Management of Doyne Honeycomb Retinal Dystrophy
Diagnosing Doyne Honeycomb Retinal Dystrophy involves a comprehensive evaluation of the patient’s medical history, symptoms, and family history of retinal diseases․ Ophthalmologic examinations, including visual acuity tests, fundoscopy, optical coherence tomography (OCT), and fluorescein angiography, are essential for assessing the presence of drusen deposits, macular changes, and potential complications․
Genetic testing plays a crucial role in confirming the diagnosis of Doyne Honeycomb Retinal Dystrophy by identifying mutations in the EFEMP1 gene associated with the condition․ Testing family members for known mutations can help assess the risk of inheritance and guide genetic counseling efforts to inform individuals about the hereditary implications of the disease․
Management of Doyne Honeycomb Retinal Dystrophy focuses on preserving existing vision, slowing disease progression, and improving quality of life for affected individuals․ Regular monitoring by ophthalmologists and low vision specialists allows for timely interventions to address visual changes and implement strategies to optimize remaining visual function․
Treatment approaches for Doyne Honeycomb Retinal Dystrophy may include lifestyle modifications to protect vision, such as wearing sunglasses to reduce light sensitivity, dietary interventions to support retinal health, and the use of low vision aids to enhance visual performance․ In some cases, anti-vascular endothelial growth factor (anti-VEGF) injections or laser therapy may be recommended to manage complications like choroidal neovascularization․
By adopting a multidisciplinary approach that integrates genetic information, ophthalmic evaluations, and personalized interventions, healthcare providers can offer holistic care to individuals with Doyne Honeycomb Retinal Dystrophy․ Research into novel treatment modalities continues to advance, providing hope for improved outcomes and quality of life for those affected by this rare inherited retinal disorder․
Challenges Faced by Individuals with Doyne Honeycomb Retinal Dystrophy
Individuals with Doyne Honeycomb Retinal Dystrophy encounter a range of challenges due to the progressive nature of the disease and its impact on vision․ One of the primary difficulties faced by affected individuals is the gradual loss of central vision, which significantly impairs tasks requiring detailed visual acuity such as reading, recognizing faces, and driving․
Reduced color perception is another common challenge experienced by individuals with Doyne Honeycomb Retinal Dystrophy․ The inability to discern colors accurately can affect daily activities like selecting clothing, coordinating home decor, and appreciating artistic works, leading to frustration and limitations in social and recreational pursuits․
Sensitivity to bright light, glare, and contrast can pose additional challenges for individuals with Doyne Honeycomb Retinal Dystrophy․ Sunlight and artificial lighting may cause discomfort and visual disturbances, making it challenging to navigate different environments and engage in outdoor activities without experiencing discomfort․
Psychological and emotional impacts are also significant challenges faced by individuals with Doyne Honeycomb Retinal Dystrophy․ Coping with vision loss, adapting to changes in daily routines, and managing uncertainties about future visual deterioration can lead to feelings of anxiety, depression, and isolation․
Furthermore, the financial burden associated with ongoing eye care, vision aids, and potential treatments for Doyne Honeycomb Retinal Dystrophy can present challenges for individuals and their families․ Access to specialized care, low vision resources, and support services is essential in addressing the multifaceted challenges faced by those living with this inherited retinal disorder․