Main Characteristics and Terminology
Osteoporosis Oculocutaneous Hypopigmentation Syndrome is a rare genetic disorder characterized by congenital oculocutaneous hypopigmentation, visual impairment, and skeletal anomalies like short stature, kyphosis, and dysmorphic facial features. The syndrome is also known as Hernández-Fragoso Syndrome (OOCHS) and has an autosomal recessive mode of inheritance. It presents with a unique pattern of severe osteoporosis alongside congenital oculocutaneous hypopigmentation. Specialists are actively involved in researching and managing this condition, with limited cases reported in the medical literature.
Main Characteristics and Terminology
Osteoporosis Oculocutaneous Hypopigmentation Syndrome is an extremely rare genetic disorder presenting with severe osteoporosis and congenital oculocutaneous hypopigmentation. It may represent a unique syndrome of unknown etiology. Three reported cases indicate an autosomal recessive mode of inheritance.
Case Study of an 8-Year-Old Male Patient
An 8-year-old male patient presented a unique pattern of congenital anomalies that included a combination of severe osteoporosis and congenital oculocutaneous hypopigmentation. The syndrome is also known as Hernández-Fragoso Syndrome (OOCHS) and has an autosomal recessive mode of inheritance. Specialists have conducted research on this rare disorder, showing that it may represent a distinct syndrome with unknown etiology.
Also Known as Hernández-Fragoso Syndrome (OOCHS)
Osteoporosis Oculocutaneous Hypopigmentation Syndrome is characterized by osteoporosis and congenital oculocutaneous hypopigmentation. It has been described as a rare genetic disorder with an autosomal recessive mode of inheritance. Three cases have been reported, shedding light on this underrepresented condition. The syndrome may involve primary bone dysplasia leading to decreased bone density.
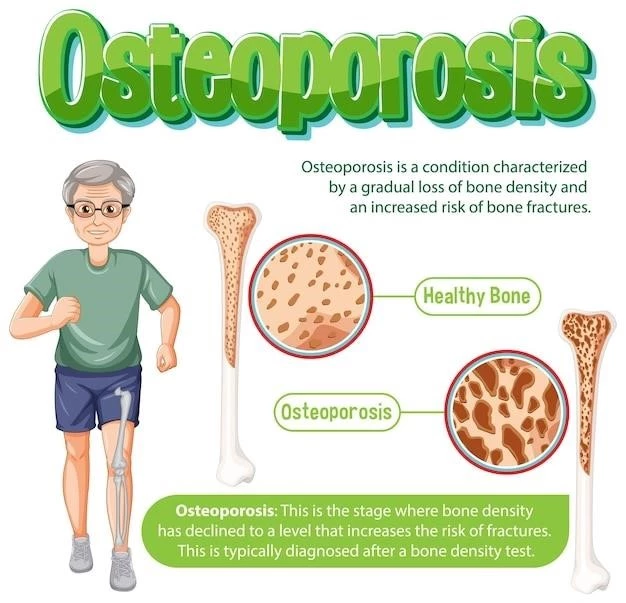
Known Synonyms and Mode of Inheritance
Also known as Hernández-Fragoso Syndrome (OOCHS)

Diagnosis and Description
Disease⁚ Osteoporosis oculocutaneous hypopigmentation syndrome is characterized by severe osteoporosis and congenital oculocutaneous hypopigmentation. Mode of inheritance is likely autosomal recessive.
Rare Genetic Disease with Congenital Oculocutaneous Hypopigmentation
The rare genetic condition Osteoporosis Oculocutaneous Hypopigmentation Syndrome is characterized by severe osteoporosis and congenital oculocutaneous hypopigmentation. This syndrome is thought to have an autosomal recessive mode of inheritance and may involve primary bone dysplasia leading to decreased bone density. Limited cases have been reported, highlighting the need for further research into this underrepresented disorder.
Osteoporosis Oculocutaneous Hypopigmentation Syndrome is believed to have an autosomal recessive mode of inheritance, based on limited cases reported in the medical literature. This rare genetic disorder is characterized by severe osteoporosis and congenital oculocutaneous hypopigmentation, presenting unique challenges in diagnosis and management.
Autosomal Recessive Nature of the Syndrome
The rare genetic disorder Osteoporosis Oculocutaneous Hypopigmentation Syndrome exhibits features of congenital oculocutaneous hypopigmentation and severe osteoporosis. Limited cases suggest an autosomal recessive inheritance pattern. Detailed analysis and further research are needed to understand this syndrome better.
Specialists are actively researching and managing cases of Osteoporosis Oculocutaneous Hypopigmentation Syndrome. These experts have received grants, written articles, conducted clinical trials, and are considered knowledgeable in this rare genetic disorder. Further investigation and collaboration with specialists are essential for advancing understanding and treatment of this condition.
Experts’ Involvement and Knowledge in Osteoporosis Oculocutaneous Hypopigmentation Syndrome
Osteoporosis Oculocutaneous Hypopigmentation Syndrome, also known as Hernández-Fragoso Syndrome (OOCHS), is a rare genetic disorder characterized by severe osteoporosis and congenital oculocutaneous hypopigmentation. Research and specialists play a crucial role in understanding and managing this syndrome. These experts receive grants, engage in clinical trials, and contribute to the knowledge base of this underrepresented condition.
Osteoporosis Oculocutaneous Hypopigmentation Syndrome presents with unique features including skeletal anomalies like short stature, visual impairment, and dysmorphic facial characteristics. Limited cases have been reported, necessitating further research into the clinical manifestations of this rare disorder.
Skeletal Anomalies, Visual Impairment, and Dysmorphic Facial Features
Osteoporosis Oculocutaneous Hypopigmentation Syndrome is characterized by unique clinical features, including skeletal anomalies like short stature, visual impairment, and dysmorphic facial characteristics. Research is crucial in understanding and managing this rare disorder.
Management strategies for Osteoporosis Oculocutaneous Hypopigmentation Syndrome involve a specialized algorithm tailored to address the unique challenges presented by severe osteoporosis and congenital oculocutaneous hypopigmentation. Comprehensive treatment options aim to mitigate the risks associated with bone fragility and visual impairment, requiring a multidisciplinary approach involving healthcare providers with expertise in managing complex genetic disorders.
Algorithm for the Management of Osteoporosis in Individuals with OOCHS
Management strategies involve a specialized algorithm tailored to address the challenges presented by severe osteoporosis and congenital oculocutaneous hypopigmentation. Treatment options aim to mitigate bone fragility risks, requiring a multidisciplinary healthcare approach.
Osteoporosis Oculocutaneous Hypopigmentation Syndrome is an extremely rare genetic disorder with fewer than 10 reported cases in the medical literature. This condition may present at birth and can affect individuals of all races and ethnicities. The syndrome’s prevalence is extremely low, making it a significant area of interest for further research and understanding.
Incidence and Affected Population of Osteoporosis Oculocutaneous Hypopigmentation Syndrome
Osteoporosis Oculocutaneous Hypopigmentation Syndrome is an extremely rare genetic disorder, with fewer than 10 cases reported in medical literature. This condition can affect individuals of any race or ethnicity, highlighting the need for further research due to its low prevalence.
Osteoporosis Oculocutaneous Hypopigmentation Syndrome presents with symptoms like bone thinning, hypopigmentation, and generalized osteoporosis. These clinical signs indicate the complex nature of this rare genetic disorder, necessitating specialized management and care.
Generalized Osteoporosis, Bone Thinning, and Hypopigmentation
Osteoporosis Oculocutaneous Hypopigmentation Syndrome is characterized by symptoms like bone thinning, hypopigmentation, and generalized osteoporosis, highlighting the complexity of this rare genetic disorder.
Osteoporosis Oculocutaneous Hypopigmentation Syndrome presents potential risks and complications due to its unique combination of severe osteoporosis and congenital oculocutaneous hypopigmentation. Individuals affected by this syndrome may face challenges related to bone fragility, vision impairment, and skeletal anomalies, necessitating specialized management and care to address these complexities.
Potential Risks and Complications Associated with the Syndrome
Osteoporosis Oculocutaneous Hypopigmentation Syndrome carries potential risks and complications due to the combined presence of severe osteoporosis and congenital oculocutaneous hypopigmentation. Patients with this syndrome may encounter challenges related to bone fragility, vision impairments, skeletal anomalies, and various dysmorphic facial features, emphasizing the need for specialized care and management.
Support networks and advocacy groups for Osteoporosis Oculocutaneous Hypopigmentation Syndrome offer assistance and information to patients dealing with this rare genetic disorder. These communities provide valuable support and resources for individuals affected by the condition, helping them navigate the complexities associated with this syndrome.
Information on Communities and Organizations Providing Assistance to Patients
Support networks and advocacy groups for Osteoporosis Oculocutaneous Hypopigmentation Syndrome offer valuable assistance and resources to individuals affected by this rare genetic disorder. These organizations aim to support and guide patients through the complexities associated with the syndrome, providing a sense of community and access to relevant information.
Clinical resources for Osteoporosis Oculocutaneous Hypopigmentation Syndrome include genetic tests, practice guidelines, and authoritative resources from various institutions and laboratories around the world. These resources provide valuable information and guidance for clinicians and researchers encountering this rare genetic disorder.
Available Genetic Tests, Practice Guidelines, and Authoritative Resources
Current clinical resources for Osteoporosis Oculocutaneous Hypopigmentation Syndrome include genetic tests, practice guidelines, and authoritative resources from specialized institutions and laboratories worldwide. These resources offer valuable insights and guidance for clinicians and researchers dealing with this rare genetic disorder.
Research efforts continue to uncover the etiology of Osteoporosis Oculocutaneous Hypopigmentation Syndrome, a rare genetic disorder with limited reported cases. Ongoing research aims to elucidate the underlying mechanisms and improve diagnostic and management strategies for this complex syndrome.
Current Understanding of the Rare Disorder and Research Efforts
Osteoporosis Oculocutaneous Hypopigmentation Syndrome is a rare genetic disorder characterized by severe osteoporosis and congenital oculocutaneous hypopigmentation. Research efforts are ongoing to elucidate the etiology of this complex syndrome and improve diagnostic and management approaches.